ATF6 and Cardiac Growth
ER stress dysregulates ER proteostasis, which activates the transcription factor, ATF6, an inducer of genes that enhance protein folding and restore proteostasis. Due to increased protein synthesis, it is possible that protein folding and, thus, ER proteostasis are challenged during cardiac myocyte growth. However, it is not known whether ATF6 is activated, and if so, what its function is during hypertrophic growth of cardiac myocytes.
In our recent studies we sought to examine the activity and function of ATF6 during cardiac hypertrophy. We found that ATF6 was activated and ATF6-target genes were induced in mice subjected to an acute model of trans-aortic constriction (TAC), or to free-wheel exercise, which promote adaptive cardiac myocyte hypertrophy with preserved cardiac function. Cardiac myocyte-specific deletion of Atf6 (ATF6 cKO) blunted TAC- and exercise-induced cardiac myocyte hypertrophy and impaired cardiac function, demonstrating a role for ATF6 in compensatory myocyte growth. Transcript profiling and chromatin immunoprecipitation identified RHEB as an ATF6-target gene in the heart. RHEB is an activator of mTORC1, a major inducer of protein synthesis and subsequent cell growth. Both TAC and exercise upregulated RHEB, activated mTORC1, and induced cardiac hypertrophy in WT mouse hearts, but not in ATF6 cKO hearts. Mechanistically, knockdown of ATF6 in neonatal rat ventricular myocytes blocked phenylephrine (PE)-, and insulin-like growth factor 1 (IGF1)-mediated Rheb induction, mTORC1 activation, and myocyte growth, all of which were restored by ectopic RHEB expression. Moreover, AAV9-RHEB restored cardiac growth to ATF6 cKO mice subjected to TAC. Finally, ATF6 induced RHEB in response to growth factors, but not in response to other activators of ATF6 that do not induce growth, indicating that ATF6 target gene induction is stress-specific.
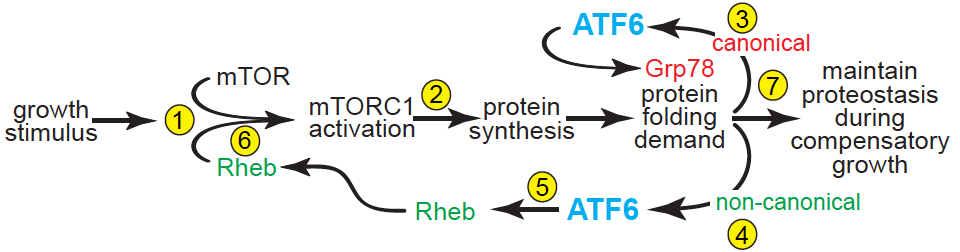
Mechanistically, this study showed that the initial event following a growth stimulus in the heart is mTORC1 activation, which depends on basal levels of Rheb (Step 1). This initial mTORC1 activation precedes, but drives initial increases in protein synthesis that place demands on the protein-folding machinery (Step 2), which activates ATF6. Then, ATF6 serves canonical- and non-canonical roles (Steps 3, 4), the latter of which includes RHEB induction (Step 5), which is necessary to sustain mTORC1 activation (Step 6) and the continued increases in protein synthesis that required for growth and cardiac myocyte hypertrophy (Step 7). From this study we concluded that compensatory cardiac hypertrophy activates ATF6, which induces Rheb and activates mTORC1. Thus, ATF6 is a previously unrecognized link between growth stimuli and mTORC1-mediated cardiac growth.